
Irreversible EGFR-TKIs: dreaming perfection
Introduction
It is hard to believe that only a decade ago the treatment of non-small-cell lung cancer (NSCLC) was based on simple exclusion of small-cell phenotype. In the last 10 years, steps toward a better knowledge of the mechanisms underlying this lethal disease moved researchers to investigate potential molecular alterations responsible for tumor growth and, consequently, for therapeutic approach. The discovery of mutations in the epidermal growth factor receptor (EGFR) has dramatically changed the treatment of NSCLC (1-3). For patients with lung adenocarcinoma and activating EGFR mutations who received first-generation EGFR-tyrosine kinase inhibitors (TKIs) - such as erlotinib or gefitinib - median overall survival (OS) ranges between 24 and 30 months (4-6), contrasting with the historical plateau of 10 months obtained with front line platinum-based chemotherapy in molecularly unselected populations (7).
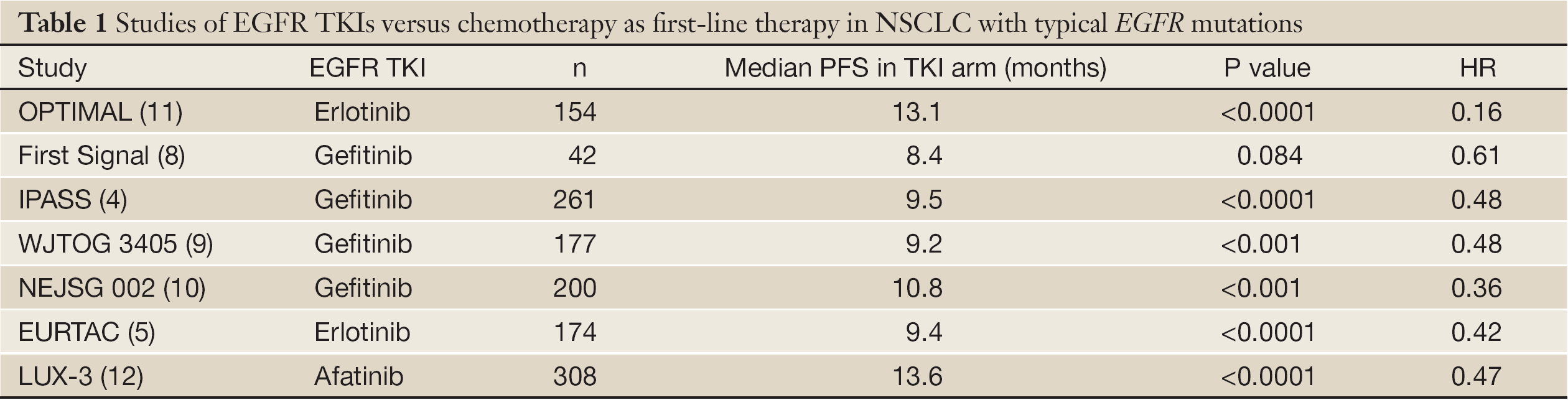
Seven large phase III randomized trials conducted in more than 1,400 patients harboring classical EGFR mutations - such as deletion in exon 19 or the L858R substitution in exon 21 - have established a new standard of care (4,5,8-12). In fact, all of these studies demonstrated the superiority of gefitinib, erlotinib or, more recently, afatinib in terms of response rate (RR) and progression free-survival (PFS) when compared to conventional platinum-doublet chemotherapy (Table 1). Because the vast majority of subjects enrolled in chemotherapy arm received an EGFR-TKIs at progression, no formal advantage in overall survival has emerged from the aforementioned trials. Nevertheless, in all trials median survival was up to 2-3 years, indicating that EGFR-TKIs are changing natural history of EGFR mutated NSCLC. Finally, since TKI toxicity is generally less severe than the one observed with platinum-based chemotherapy, offering an EGFR-TKIs to a sensitive patient means delay toxic effects of chemotherapy and preserve quality of life (QoL). Similarly, a significant benefit was observed in those EGFR mutant patients treated with erlotinib or gefitinib as second- or third-line treatment (13,14) as well as in maintenance setting (15,16). Taken into account, all these data reinforced the conviction that patients carrying an activating EGFR mutation should never loose the opportunity of receiving an EGFR-TKI during the course of their disease.

Full table
However, the enthusiasm generated by these findings has been modulated by the awareness that, until now, no patient can be cured and inevitably all our patients progress and die for their disease. Aim of the present article is to briefly discuss the pitfalls of the first generation EGFR TKIs and to highlight the available data on a new class of inhibitors, also called irreversible or covalent, in the treatment of NSCLC.
Unmet needs with reversible EGFR-TKIs
Main criticisms related to first-generation EGFR-TKIs are listed in Table 2.

Full table
First, a consistent proportion of EGFR mutant patients, approximately 30%, never respond to anti-EGFR TKIs due to primary resistance and the mechanism of this phenomenon is poorly understood (17). On the other hand, we know that EGFR mutation does not mean sensitive mutation. EGFR mutations exist in exon 18-21 of the tyrosine-binding domain of the EGFR (1,2,18). As previously reported, deletion in exon 19 and L858R point mutation in exon 21 account for the 90% of EGFR mutations detected in NSCLC and are clearly associated with benefit to EGFR TKIs (4,5,8-12). Beside these classical or typical mutations, there is still a small group of “uncommon” mutations, as G719, S768, L861 and others, that can occur with or without a common mutation (19) and for which the clinical impact is poorly understood. Wu et al., analyzed a large series of 1,261 lung cancer cases of which 627 were EGFR mutant, with the aim to evaluate the outcome to erlotinib or gefitinib according to the type of mutation (20). The authors confirmed that typical mutations derived the greatest benefit in terms of RR, PFS and OS (74%, 8.5 and 19.6 months respectively) from such treatment; nevertheless the absolute difference in outcome was not so huge when considering the less frequent G719 and L861 mutations (RR 53.3% and 60.0%, PFS 8.1 and 6.0 months, OS 16.4 and 15.2 months for G719 and L861 respectively); on the other hand, some rare uncommon mutations (i.e., V769M and A871E) failed to respond to EGFR TKIs (RR 20%, PFS 1.6 months and OS 11.1 months) with a clinical trend that was very similar to that observed for EGFR wild type population (RR 16.5%, PFS 2.0 months and OS 10.4). Although, the retrospective nature of the investigation and the low sample size of uncommon mutations in large phase III trials, only 6% and 3.8% in the NEJ002 and IPASS respectively (4,10), do not permit to drawn any definitive conclusion, at the present time it is not recommended in clinical practice to treat in first-line a patient with uncommon mutation with erlotinib or gefitinib.
Second, treatment with reversible EGFR TKIs is generally defined as “overall well tolerated”. Indeed in the large phase III trials comparing erlotinib and gefitinib versus standard platinum based chemotherapy, also the toxicity profile was significantly better in the “experimental” arms; the incidences of grade >3 skin rash, diarrhea and liver dysfunction, the three most common adverse events related to EGFR TKIs treatment, did not exceed 20% and the proportion of patients that discontinued therapy due to toxic effects is less than 10% (4,5,8-10). Nevertheless, this small amount of patients, even if molecularly-favored, no longer benefited from therapy. On the other hand, unlike conventional chemotherapy, treatment with targeted agents is continued until disease progression; as a consequence also a long-lasting grade 2 toxicity could became “psicologically serious” over the time mainly because, more often, treated patients are young and able to normal activities.
Last but not least, the most relevant problem related to EGFR TKI therapy is the emergence of acquired resistance (21-23). Indeed, despite an initial dramatic tumor regression in up to 80% of cases after a median time of 9-12 months, all patients’ progress and the possibility of further control tumor growth inevitably decreases.
Acquired resistance to EGFR TKIs: clinical, biological and therapeutic implications
From a clinical point of view, we refer to acquired resistance according to the criteria proposed by Jackman and coworkers (24) in 2010 considering as “resistant” those patients treated with single-agent erlotinib or gefitinib (I) who progressed while on treatment and (II) who harbored a sensitive EGFR mutation or (III) if EGFR status is wild type or unknown, who obtained partial or complete response or a significant and durable (>6 months) clinical benefit - according to RECIST or WHO criteria - after initiation of EGFR TKI therapy. Two important issues derived from this work: first, the utility of a relative simple criteria to correctly define and select for novel clinical trials a population otherwise too heterogeneous; second, the concept that a progression that occur while on treatment could be interpreted as a transitory clinical condition related to the type of therapy (i.e., reversible EGFR TKIs) rather than to a true EGFR-pathway-independent tumor growth. In other words, the sensitivity to an anti-EGFR TKIs could be restore after a break period (3,22,25); for this reason many trials with sequential use of chemo- and EGFR targeted therapies are ongoing (25).
From biological point of view, prolonged exposure to erlotinib or gefitinib provides selective pressure for the development of tumor clones able to growth irrespective of the drug inhibition. The mechanisms underlying the phenomenon of secondary resistance are object of extensive evaluation and some of these are so far elucidated (22,23,26). Several preclinical studies demonstrated that the two main mechanisms responsible for acquired resistance are the up-regulation of the downstream signal by mesenchymal-epidermal transition (MET) amplification and the emergence of T790M EGFR gatekeeper mutation (26-30). Other mechanisms include EGFR amplifications, PI3KCA mutations or a transition from epiyhelial to mesenchymal differentiation (26). More interestingly, for a little percentage of resistant tumors occurs transformation into SCLC (26).
MET amplification is found to be associated with acquired resistance in up to 20% of cases and inhibition of MET with the use of monoclonal antibodies (31-33) or small molecule TK inhibitor (34) alone or in combination with other targeted agents are currently under investigations. Anti-MET strategies have been extensively discussed elsewhere (35-37).
The “acquired” T790M mutation - a characteristic point mutation in the exon 20 of the EGFR gene - is associated with lack of activity of first generation EGFR TKI and is responsible for secondary resistance in at least 50% of patients exposed to erlotinib or gefitinib (22,23,26,38). Initial data showed that this event occur in less than 3% of mutated patients before starting and EGFR TKI therapy (30). More recently, using high sensitive methods, the EGFR T790M mutation was detected in up to 40% of previously untreated NSCLC, suggesting that what we call an “acquired resistance” is a pre-existing phenomenon (39). Retrospective data from Memorial Sloan Kettering Cancer Center suggested that this molecular event is largely underestimated, when assessed by low-sensitive technique (39). Whereas the vast majority of EGFR mutations are sensitive to TKIs because they decrease the affinity of the receptor for its natural substrate ATP, the presence of T790M, altering the conformation of the tyrosine kinase domain of the EGFR, restore its affinity for ATP at the levels similar than reported for EGFR wild type thus reducing the ability of reversible TKIs to effectively compete with ATP (40-41). In vitro studies demonstrated that gefitinib-resistant as well T790M mutation positive clones remain sensitive to irreversible EGFR TKIs that are structurally similar to erlotinib and gefitinib (42); unlike reversible TKIs, this new class of inhibitor contain an acceptor-group that binds covalently with the Cys797 present at the ATP-binding site of mutant EGFR. As discussed above, due to their characteristics irreversible EGFR TKIs seemed to be the ideal compounds to test in order to overcome T790M acquired resistance (42).
A fascinating way to interfere with the signaling cascade of the EGFR, in order to overcome resistance, is to simultaneously inhibit both the extracellular and intracellular receptor domains. The clinical proof of the so-called “vertical inhibition” comes from previous experience in HER2-overexpressing trastuzumab-resistant metastatic breast cancer, in which the combination of trastuzumab and lapatinib was superior to lapatinib alone in terms of RR and PFS (43).
Similarly in NSCLC, the combination of afatinib and cetuximab induced nearly complete tumor regression in T790M transgenic murine models (44). On this base, a pivotal phase Ib study has been recently conducted in NSCLC patients with clinically defined acquired resistance with the aim to explore the safety and activity of the combination (45). In the initial cohort, 22 patients were exposed to afatinib at the oral daily dose of 40 mg and cetuximab 500 mg/m2 intravenously every 2 weeks. Adverse events were consistent with the typical class-effects previously reported (i.e., diarrhea and skin rash) and were generally mild, with only 3 patients experiencing grade 3 skin toxicity. Every patient obtained disease control with a median reduction in tumor size of 76% and a promising activity of 36% (8/22 including 4/13 T790M positive cases), leading to enrollment of an additional cohort of 80 patients. Final results have been recently presented. Main grade 3 adverse events were skin rash (12%) and diarrhea (6%); 96 patients were evaluable for efficacy and treatment resulted in 75% of disease control rate with a response rate of 30%, without significant difference between T790M positive and T790M negative patients (32% versus 28% months); median PFS was 4.7 months (46). These encouraging results deserve further validation in large phase III trials.
New generations EGFR TKIs
The second generation of EGFR inhibitors, also-defined irreversible or covalent EGFR inhibitors, afatinib, dacomitinib and neratinib, are pan-ErbB inhibitors and their activity against both EGFR activating mutations and the T790M mutation has been demonstrated in in vivo models (47-49).
Afatinib
Afatinib (BIBW2992) binds irreversibly to EGFR, HER2, HER4 and also to EGFR receptors carrying the T790M mutation, suggesting a potential role in overcoming resistance. Multiple phase I studies identified in 50 mg once daily the maximum tolerated dose (MTD) with main toxicities represented by diarrhea and skin rash (50). On this basis, the LUX-Lung clinical trial program has been launched for testing this molecule in different setting in advanced NSCLC patients.
In the phase 2b/3 LUX-Lung 1 trial (51), a total of 585 adenocarcinoma patients who met criteria for acquired resistance to EGFR-TKIs as proposed by Jackman et al. (24), were randomized in a 2:1 fashion to receive daily oral afatinib 50 mg plus best supportive care (BSC) or placebo plus BSC as third or subsequent line of therapy. The primary end-point was overall survival. Interestingly, the trial did not need archival tumor tissue and the subjects were not screened for EGFR status, but the prior disease control for >3 months under TKIs treatment was used as surrogate criterion to increase probability of EGFR mutations. The treatment with afatinib resulted in better activity (RR 7% versus 0.5%) and longer PFS (3.3 months, 95% CI, 2.79-4.40 months) than it was in placebo group (1.1 months, 95% CI, 0.95-1.68 months, HR 0.38, P<0.0001). Surprisingly, the PFS benefit did not translate in survival benefit. Median overall survival was 10 and 12 months for the afatinib and placebo arm respectively; the reason behind this unusual finding could be the confounding effect of post-study therapies; indeed, a greater proportion in the placebo arm than in the afatinib arm receive subsequent treatment, including chemotherapy and EGFR TKI.
Similar activity was preliminary reported in the LUX-Lung 4, a phase II open label trial, in which 62 Japanese patients who progressed after 1 or 2 chemotherapy lines and prior erlotinib or gefitinib underwent therapy with afatinib at the dose 50 mg (52). Response rate was 8%, with DCR of 66%, while PFS resulted of 4.4 months.
Afatinib was also evaluated as first line and second line therapy in patients who had not received a first generation TKI. The LUX-Lung 2 trial was a single-arm, multicenter phase II study evaluating the efficacy of afatinib 40-50 mg daily in advanced adenocarcinoma with EGFR activating mutations (53). A total of 129 subjects (first line N=61; second line, N=68) were enrolled onto the study; notably 18% of patients presented an uncommon mutation. In overall population objective RR, DCR and PFS were 59%, 83% and 14 months respectively, with a median overall survival of 24 months; no difference in outcome was noted between patients harbored L858R or deletion in exon 19 irrespective of line of therapy, while the efficacy in terms of RR, PFS and OS was lower in those patients with uncommon mutations (RR 39%; median PFS 3.7 months; OS 16.3 months).
The LUX-lung 3, the first phase III study using the combination of pemetrexed and cisplatin as a comparator arm, randomly assigned in a 2:1 fashion EGFR mutant adenocarcinoma patients to receive as front line therapy afatinib 40 mg daily or six cycles of chemotherapy (12). The study, which enrolled 345 patients, met its primary end point of PFS. Patients treated with afatinib had a 42% relative reduction in risk of progression compared with those receiving standard chemotherapy (11.1 versus 6.9 months, HR 0.58; 13.1 versus 6.9 months, HR 0.47 for patients with classical EGFR mutations). Treatment with afatinib was also associated with higher response rate (56% versus 23%, ITT population) and better toxicity profile than chemotherapy, although G3 diarrhea and skin rash occurred in 14% and 16% of cases receiving the study drug.
Dacomitinib
Dacomitinib (PF0299804), covalently binds the adenosine triphosphate domain of each of three kinase active members of the HER family: EGFR/HER1, HER2 and HER4. In preclinical experiences, dacomitinib showed greater antitumor activity in gefitinib-resistant NSCLC in vitro and in vivo models (49). In NSCLC clinical trials, Dacomitinib has been evaluated in three different setting: after EGFR TKI failure (54-56), in second line in patients not previously exposed to a reversible EGFR TKI and in front line in EGFR mutants patients (57,58).
In a phase I study (54), a disease control rate (PR + SD) of 34% was seen in 44 patients pretreated with first-generation EGFR TKIs (94%) and chemotherapy (79%); most frequently any-grade adverse events observed at the recommended daily dose of 45 mg were diarrhea (78%) and skin rash (65%). In another phase I/II trial conducted in 36 advanced NSCLC patients who progressed after one or two prior chemotherapy regimen and erlotinib (55), DCR was observed in 67% and 40% of patients with adenocarcinoma and squamous cell carcinoma respectively. In another Korean phase II trial (56), enrolling 42 patients with similar characteristics, preliminary results demonstrated an activity of 15% with a DCR of 25%.
Ramalingam et al. published the results of the first randomized trial on irreversible EGFR TKI in lung cancer patients never exposed to TKI treatment (59). Subjects enrolled onto this phase II study were randomly assigned to receive as second line treatment erlotinib (N=94) or dacomitinib (N=94). The primary end point was PFS. In the dacomitinib arm there was a higher number of patients with ECOG performance status 2, EGFR mutant and treated with 2 or more prior chemotherapy than in the erlotinib arm. PFS resulted in favor of the experimental arm (median PFS 2.8 versus 1.91 months; HR 0.66); the improvement in PFS was reported across most of the subgroup considered and particularly in KRAS wild type/EGFR any status (median PFS 3.71 versus 1.91 months; HR 0.55), KRAS wild type/EGFR wild type (median PFS 2.21 versus 1.84 months; HR 0.61), while for EGFR mutant patients median PFS resulted of 7.44 in both arms. The objective RR was lower in the erlotinib arm than in dacomitinib arm (5.3% versus 17%), as DCR (14.9% versus 29.8%) did. However, grade diarrhea and skin rash were more frequent with dacomitinib than with erlotinib.
More recently, Kris et al. reported the results of the 1017 study of dacomitinib at the dose of 30-45 mg daily in NSCLC patients with EGFR mutations or HER-2 mutations (i.e., exon 20 insertions or point mutations) or HER-2 amplification (57). Endpoints included progression-free survival rate at 4 months (PFS at 4 M), PFS, partial response (PR) rate and safety. EGFR cohort included never or light-former smoker (<10 pack year) patients with metastatic non-pretreated adenocarcinoma or treatment-naïve patients with known EGFR mutations, while HER2 cohort enrolled subjects with HER2 mutations or amplification who received any number of prior therapy. In the EGFR cohort (Cohort A, N=89), 46 of patients harbored a classical mutation (exon 19, N=25; exon 21, N=21); in this subgroup, RR rate was 76% while PFS at 4M and PFS were 95.5% (95% CI, 83.2-98.9%) and 18.2 months (95% CI, 12.8-23.8 months) respectively. As expected, common side effects were diarrhea, skin toxicity and nail changes. Cohort B is still recruiting and in the first 22 enrolled patients (HER2 amplification, N=4; HER2 mutation, N=18) an interesting activity of 14% was observed, but limited to those patients carrying a HER-2 mutation.
Neratinib
Neratinib (HKI-272), an irreversible HER family inhibitor targeting EGFR/HER-1, HER-2 and HER-4, was initially tested in a phase I trial of 72 patients with advanced ErbB2 or ErbB1/EGFR IHC positive tumors (58). Maximum tolerated dose (MTD) was determined to be 320 mg and the most common related adverse event at this dose was diarrhea. Strikingly, a long-lasting disease control (defined as stable disease for >24 weeks) was observed in 43% of refractory NSCLC patients.
A large non-randomized phase II tria l explored the activity of neratinib in three different cohorts of advanced pretreated NSCLC patients (60). Arm A included patients with activating EGFR mutation (N=91), arm B included EGFR wild-type patients (N=48) while arm C included EGFR TKI-naïve patients selected for adenocarcinoma histology and smoking history (N=28). Subjects in arms A and B had to have received at least 12 weeks of prior erlotinib/gefitinib treatment. In the overall population (N=158), the activity was lower than expected, with only 2% of responders (RR 3.4% arm A; 0% arm B; 0% arm C). Interestingly, the three responding patients harbored the rare G719X point mutation in exon 18, maybe suggesting that neratinib could be less effective in presence of classical EGFR mutations; on the contrary, the presence of T790M mutation did not seem guarantee any benefit from such treatment. Median PFS was 15.3 weeks in the entire cohort, without significant difference between the three arms (15.3, 16.1 and 9.3 weeks in arm A, B and C respectively). Nevertheless, in the first 39 patients receiving neratinib at the dose of 320 mg daily the occurrence of grade 3 diarrhea was unacceptably high (50%); as a consequence, a dose reduction to 240 mg was required in order to improve tolerability with the hypothetical disadvantage of negatively affect response. Anyway, this major limitation led to dissipate the interest to further explore neratinib in NSCLC.
Discussion
The ideal inhibitor might be equally effective irrespective of the type of EGFR mutations, highly similar to the binding site of the receptor, active even in presence of T790M clones and - from the patient point of view - at least with identical or better toxicity profile than older compounds. Have the irreversible EGFR TKIs met all this endpoints?
In front line setting, the efficacy of covalent inhibitors is comparable to the one reported for reversible TKIs. In the LUX Lung 3 trial median PFS for patients with typical EGFR mutations is more than 13 months, with an absolute improvement of nearly 7 months respect to chemotherapy arm (12). These results is quite similar to those reported in the OPTIMAL trial, in which an impressive HR of 0.16 for PFS in favour of erlotinib arm was observed (11); nevertheless, unlike OPTIMAL, in the LUX-3 the difference in outcome between EGFR-TKI therapy and chemotherapy appears to be real, considering the high performance of the comparator arm. In phase II trial, Dacomitinib showed an unexpected PFS of nearly 18 months, but this finding deserves further validation in prospective large phase III studies (57). In terms of activity, best response rate observed in phase II trials of first and second generation EGFR-TKIs seemed almost identical for both class of inhibitors (53,57,61,62) (Table 3). Large phase III trials comparing head-to-head irreversible versus reversible EGFR TKIs are urgently needed to define whether covalent inhibitors may improve outcomes and possibly delay the onset of resistance.

Full table
Once again, patients harboring a classical mutation gained the greatest benefit from such treatments. In the LUX Lung 2, in which 18% of patients presented uncommon mutations, the RR and PFS was lower for this population and in any case, were consistent with those reported for gefitinib and erlotinib (53). In the LUX Lung 3 study (12,63), 48 (10.6%) patients presented uncommon mutations that were were categorized into 5 groups: T790M, G719X, S768I, exon 20 insertions, L861Q; the first 3 groups included double mutant patients. Tumour response and prolonged PFS were noted in 2 double mutant patients (L858R + T790M; S768I + L858R) and in 2 with single uncommon mutation (G719X and S768I), while in the other cases SD was the best response. Nevertheless these results are inconclusive, as the effect of afatinib in doublet mutant patients could be in part referred to the presence of the L858R mutation. As previously reported (60), neratinib seemed to be more effective in presence of the rare G719X mutation; this might simply reflect a different sensitivity of specific mutations to an EGFR TKI. Furthermore, is it not possible to exclude that this result was obtained by chance because of the very small number of patients.
Irreversible TKIs have been developed with a specific focus on patients with acquired resistance to erlotinib or gefitinib. LUX-Lung 1 (51) and LUX-Lung 4 (52) trials failed to demonstrate a clear benefit in terms of RR in patients with acquired resistance and particularly in those cancers with T790M; the activity reported in the 2 studies was only 7% and 8%, lower than expected. We recently presented a retrospective analysis of 68 advanced lung adenocarcinoma patients with acquired resistance to reversible EGFR TKIs treated with afatinib and we reported a response rate of 10.6% with a disease control rate of 65%. Four of the five responding patients harbored a classical mutation including 1 patient with T790M; in 9 patients in which tumor biopsy was repeated before starting afatinib, only 2 patients had T790M mutation, with no evidence of response (64). All these results are disappointing and suggest that the ability of covalent inhibitor in overcome acquired resistance may have limitations unpredicted in preclinical experiences; a possible explanation could be the different drug concentration achieved in humans respect to preclinical models.
Another critical issue concerns the toxicity profile of the irreversible inhibitors. In metastatic setting, the preservation of QoL still remains one of the goals of therapy, mainly when considering second and subsequent line of treatment. In the case of neratinib, an unacceptable incidence of 50% of grade diarrhea required a dose reduction in the Sequist’s phase II trial (60). Grade 3 adverse events reported in LUX 1 and 2 trials (51,52), led the clinicians to consider 40 mg as the “optimal” tolerated dose, instead of 50 mg defined in phase I trial (50). Anyway, indirect comparison of phase III trials showed higher incidences of diarrhea, skin rash and stomatitis for afatinib respect to erlotinib or gefitinib (4,5,8). Main grade >3 toxicities with EGFR-TKIs are listed in Table 4. Taken into account, all these data suggested that toxicities of covalent inhibitors are probably higher than those observed with first-generation compounds.

Full table
Conclusions
Irreversible EGFR TKIs could represent a promising therapeutic option in the treatment of NSCLC. Although in absence of trials directly comparing reversible versus irreversible TKIs, available data failed to demonstrated a superior efficacy respect to first-generation inhibitors. Furthermore, the activity reported in patients harbouring an EGFR uncommon mutation is consistent with the one observed for gefitinib and erlotinib. Although the clinical development of covalent inhibitors focused on T790M-dependent acquired resistance, activity observed in this particular subgroup was only modest. The high affinity for ATP binding site could in part explain the prevalence of typical class-effects observed with afatinib, neratinib and dacomitinib. Results from ongoing and planned clinical trials, will help us to define the role of second generation TKIs in our clinical practice.
Acknowledgements
Disclosure: The authors declare no conflict of interest.
References
- Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497-500.
- Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004;101:13306-11.
- Soria JC, Mok TS, Cappuzzo F, et al. EGFR-mutated oncogene-addicted non-small cell lung cancer: current trends and future prospects. Cancer Treat Rev 2012;38:416-30.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947-57.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239-46.
- Mok TS, Lee JS, Zhang L, et al. Biomarkers analyses and overall survival (OS) from the randomized, placebocontrolled, phase 3, FASTACT-2 study of intercalated erlotinib with first- line chemotherapy in advanced nonsmall cell lung cancer (NSCLC). Ann Oncol 2012;29:ix 400;abstr 12260.
- Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-smallcell lung cancer. N Engl J Med 2002;346:92-8.
- Han JY, Park K, Kim SW, et al. First-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J Clin Oncol 2012;30:1122-8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121-8.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010;362:2380-8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735-42.
- Yang JC, Schuler MH, Yamamoto N, et al. LUX-Lung 3: A randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations. J Clin Oncol 2012;30:abstr LBA7500.
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005;353:123-32.
- Douillard JY, Shepherd FA, Hirsh V, et al. Molecular predictors of outcome with gefitinib and docetaxel in previously treated non-small-cell lung cancer: data from the randomized phase III INTEREST trial. J Clin Oncol 2010;28:744-52.
- Cappuzzo F, Ciuleanu T, Stelmakh L, et al. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: a multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol 2010;11:521-9.
- Zhang L, Ma S, Song X, et al. Gefitinib versus placebo as maintenance therapy in patients with locally advanced or metastatic non-small-cell lung cancer (INFORM; C-TONG 0804): a multicentre, double-blind randomised phase 3 trial. Lancet Oncol 2012;13:466-75.
- Cappuzzo F, Jänne PA, Skokan M, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol 2009;20:298-304.
- Riely GJ, Politi KA, Miller VA, et al. Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin Cancer Res 2006;12:7232-41.
- Chen Z, Feng J, Saldivar JS, et al. EGFR somatic doublets in lung cancer are frequent and generally arise from a pair of driver mutations uncommonly seen as singlet mutations: one-third of doublets occur at five pairs of amino acids. Oncogene 2008;27:4336-43.
- Wu JY, Yu CJ, Chang YC, et al. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res 2011;17:3812-21.
- Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005;352:786-92.
- Brugger W, Thomas M. EGFR-TKI resistant nonsmall cell lung cancer (NSCLC): new developments and implications for future treatment. Lung Cancer 2012;77:2-8.
- Engelman JA, Jänne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 2008;14:2895-9.
- Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol 2010;28:357-60.
- Moran T, Sequist LV. Timing of epidermal growth factor receptor tyrosine kinase inhibitor therapy in patients with lung cancer with EGFR mutations. J Clin Oncol 2012;30:3330-6.
- Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26.
- Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039-43.
- Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007;104:20932-7.
- Chen HJ, Mok TS, Chen ZH, et al. Clinicopathologic and molecular features of epidermal growth factor receptor T790M mutation and c-MET amplification in tyrosine kinase inhibitor-resistant Chinese non-small cell lung cancer. Pathol Oncol Res 2009;15:651-8.
- Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005;2:e73.
- Tan E, Park K, Lim WT, et al. Phase Ib study of ficlatuzumab (formerly AV-299), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb) in combination with gefitinib (G) in Asian patients (pts) with NSCLC. J Clin Oncol 2011;29:abstr 7571.
- Mok TS, Park K, Geater SL, et al. A Randomized Phase 2 Study with Exploratory Biomarker Analysis of Ficlatuzumab, a Humanized Hepatocyte Growth Factor (HGF) Inhibitory Monoclonal Antibody, in Combination with Gefitinib versus Gefitinib Alone in Asian Patients With Lung Adenocarcinoma. Ann Oncol 2012;23:abstr.
- Spigel DR, Ervin TJ, Ramlau R, et al. Final efficacy results from OAM4558g, a randomized phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLC. J Clin Oncol 2011;29:abstr 7505.
- Sequist LV, von Pawel J, Garmey EG, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol 2011;29:3307-15.
- Blumenschein GR Jr, Mills GB, Gonzalez-Angulo AM. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J Clin Oncol 2012;30:3287-96.
- Appleman LJ. MET signaling pathway: a rational target for cancer therapy. J Clin Oncol 2011;29:4837-8.
- Toschi L, Cappuzzo F. Clinical implications of MET gene copy number in lung cancer. Future Oncol 2010;6:239-47.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFRmutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616-22.
- Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res 2011;17:1169-80.
- Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070-5.
- Suda K, Onozato R, Yatabe Y, et al. EGFR T790M mutation: a double role in lung cancer cell survival? J Thorac Oncol 2009;4:1-4.
- Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A 2005;102:7665-70.
- Blackwell KL, Burstein HJ, Storniolo AM, et al. Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol 2010;28:1124-30.
- Regales L, Gong Y, Shen R, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest 2009;119:3000-10.
- Janjigian YY, Groen HJ, Horn L, et al. Activity and tolerability of afatinib (BIBW2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. J Clin Oncol 2011; 29:abstr 7525.
- Janjigian YY, Smith EE, Horn L, et al. Activity of afatinib/ cetuximab in patients with EGFR mutant non-small cell lung cancer and acquired resistance to EGFR inhibitors. Ann Oncol 2012;23:ix 401.
- Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008;27:4702-11.
- Ninomiya T, Takigawa N, Kubo T, et al. Effect of afatinib on lung cancer burden induced by an exon 19 EGFR mutation in transgenic mice [abstract 3566]. Presented at the 102nd Annual Meeting of the American Association for Cancer Research; Orlando, Florida; April 2-11, 2011.
- Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 2007;67:11924-32.
- Yap TA, Vidal L, Adam J, et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J Clin Oncol 2010;28:3965-72.
- Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic nonsmall- cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUXLung 1): a phase 2b/3 randomised trial. Lancet Oncol 2012;13:528-38.
- Atagi S, Katakami N, Hida T, et al. LUX Lung 4: a phase II of Afatinib (BIBW2992) in advanced NSCLC patients previously treated with erlotinib or gefitinib. J Thor Oncol 2011;6:abstr O19.06.
- Yang JC, Shih JY, Su WC, et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trial. Lancet Oncol 2012;13:539-48.
- Janne PA, Schellenns JH, Engelman JA, et al. Preliminary activity and safety results from a phase I clinical trial of PF-00299804, an Irreversible pan-HER inhibitor in patients with NSCLC. J Clin Oncol 2008;26:abstr 8027.
- Janne PA, Reckamp K, Koczywas M, et al. A phase 2 trial of PF-00299804, an oral irreversible HER kinase inhibitor (TKI) in patients with advanced NSCLC after failure of prior chemotherapy: preliminary efficacy and safety results. J Thorac Oncol 2009;4:S293-94.
- Park K, Heo DS, Cho BC, et al. PF299804 in Asian patients with non-small-cell lung cancer refractory to chemotherapy and erlotinib or gefitinib: aphase I/II study. J Clin Oncol 2010;28:abstr 7599.
- Kris MG, Mok T, Ou SH, et al. Dacomitinib (PF- 00299804), an Irreversible pan-HER Tyrosine Kinase Inhibitor, for First-Line Treatment of EGFR-Mutant or HER2-Mutant or -Amplified Lung Cancers. J Clin Oncol 2012;30:abstr 7530.
- Wong KK, Fracasso PM, Bukowski RM, et al. A phase I study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res 2009;15:2552-8.
- Ramalingam SS, Blackhall F, Krzakowski M, et al. Randomized phase II study of dacomitinib (PF- 00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol 2012;30:3337-44.
- Sequist LV, Besse B, Lynch TJ, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced nonsmall- cell lung cancer. J Clin Oncol 2010;28:3076-83.
- Jänne PA, Wang X, Socinski MA, et al. Randomized phase II trial of erlotinib alone or with carboplatin and paclitaxel in patients who were never or light former smokers with advanced lung adenocarcinoma: CALGB 30406 trial. J Clin Oncol 2012;30:2063-9.
- Sequist LV, Martins RG, Spigel D, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol 2008;26:2442-9.
- Yang JC, Schuler M, Yamamoto N, et al. Activity of afatinib in uncommon epidermal growth factor receptor (EGFR) mutations in LUX-Lung 3, a phase III trial of afatinib or cisplatin/pemetrexed in EGFR mutationpositive lung cancer. Ann Oncol 2012;23:ix410, abstr 1256p.
- Landi L, galetta D, Bennati C, et al. Efficacy of the irreversible EGFR-HER2 dual inhibitor afatinib in pretreated lung adenocarcinoma. Ann Oncol 2012;23:ix423, abstr 1288P.


{kind=link}